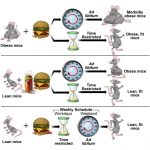
Inflammatory process in Alzheimer’s Disease
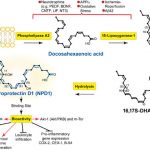
Contents
Alzheimer Disease
Alzheimer Disease (AD) is a neurodegenerative disorder and the most common form of dementia. Histopathologically is characterized by the presence of two major hallmarks, the intracellular neurofibrillary tangles (NFTs) and extracellular neuritic plaques (NPs) surrounded by activated astrocytes and microglia. NFTs consist of paired helical filaments of truncated tau protein that is abnormally hyperphosphorylated. The main component in the NP is the amyloid-β peptide (Aβ), a small fragment of 40–42 amino acids with a molecular weight of 4 kD. It has been proposed that the amyloid aggregates and microglia activation are able to favor the neurodegenerative process observed in AD patients. However, the role of inflammation in AD is controversial, because in early stages the inflammation could have a beneficial role in the pathology, since it has been thought that the microglia and astrocytes activated could be involved in Aβ clearance. Nevertheless the chronic activation of the microglia has been related with an increase of Aβ and possibly with tau phosphorylation. Studies in AD brains have shown an upregulation of complement molecules, pro-inflammatory cytokines, acute phase reactants and other inflammatory mediators that could contribute with the neurodegenerative process. Clinical trials and animal models with non-steroidal anti-inflammatory drugs (NSAIDs) indicate that these drugs may decrease the risk of developing AD and apparently reduce Aβ deposition. Finally, further studies are needed to determine whether treatment with anti-inflammatory strategies, may decrease the neurodegenerative process that affects these patients.
NPs, are extracellular deposits structures that contain a highly insoluble fibrillar Aβ core formed by fragments of 39–42 amino acids surrounded by microglia, reactive astrocytes, and dystrophic neurites produced from degenerating neuronal processes (Iversen et al., 1995). Aβ normally originates from APP-β (Kang et al., 1987) through the sequential action of beta secretase and the multi-protein gamma-secretase complex.
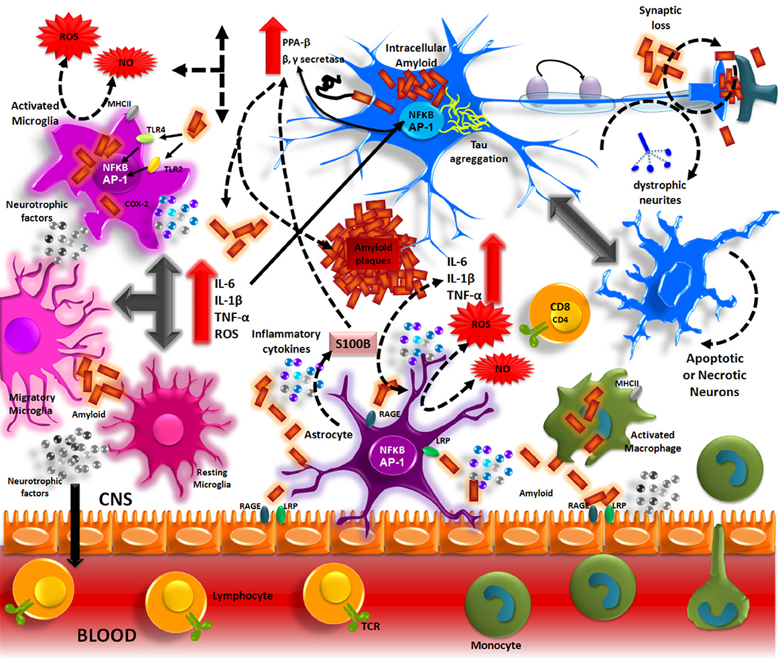
Aβ accumulation in the brain, in one of the main pathological processes of AD patients. The formation of these deposits initiates a series of cellular events which are able to elicit an immune response where resident cells such as microglia and astrocytes could participate. Aβ accumulation in parenchyma and blood vessels causes microglial migration and promotes acute and chronic inflammatory responses against the aggregates, thus inducing the production of nitric oxide (NO), reactive oxygen species (ROS), pro-inflammatory cytokines (TNFα, IL-1β and IL-6), and PGs (PGE2), which eventually could promote neuronal death (Figure 1) (Akiyama et al., 2000; Kitazawa et al., 2004).
Inflammation in Alzheimer’s disease.
The Aβ peptide produced by APP processing, form aggregates that activate microglia through TLRs and RAGE receptors. These receptors in turn, activate NF-κ B and AP-1 transcription factors, which induce the reactive oxygen species (ROS) production and the expression of inflammatory cytokines (IL-1, IL-6, TNF). These inflammatory factors directly acting on the neurons and also stimulate the astrocytes, which amplify the pro-inflammatory signals, inducing a neurotoxic effects. The inflammatory mediators generate by resident CNS cells, induce the production of adhesion molecules and chemokines, which recruit peripheral immune cells.
Nitric Oxide
NO is a molecule that contributes importantly in cell signaling. In the presence of oxygen, L-arginine, is converted to L-citrulline and release NO. Nitric oxide synthase (NOS) is the enzyme responsible for catalyzing this reaction, and it occurs in three isoforms. In the CNS, the neuronal isoform (nNOS) is widely distributed in neurons, astrocytes and blood vessels. The endothelial isoform (eNOS) is located in the hippocampal pyramidal neurons, endothelial cells and some astrocytes. The expression of the inducible isoform (iNOS) is typically low but is increased in microglia and astrocytes during neuro-inflammation. Under physiological conditions, it is believed that NO regulates the release of neurotransmitters and hormones and promotes cell survival and long-term potentiation. However, high levels of NO are generated in inflammatory conditions, which might contribute to synaptic transmission dysfunction, protein and lipid oxidative damage, excitotoxicity, and neuronal death (Liu et al., 2002; Bishop and Anderson, 2005; Calabrese et al., 2007).
Tissue and neuronal analyses of AD patients have revealed that Aβ is able to promote NOS expression and NO production in microglia cells and reactive astrocytes (Goodwin et al., 1997; Wallace et al., 1997; Akama et al., 1998). Aβ also promotes the pro-inflammatory cytokines IL-1β and TNF-α liberation which contribute to NO and peroxynitrite formation (Rossi and Bianchini, 1996; Combs et al., 2001) and cause protein and lipid modifications, mitochondrial damage, apoptosis and promote Aβ formation, increasing the γ-secretase complex activity (Torreilles et al., 1999; Keil et al., 2004; Guix et al., 2012). Under normal conditions, NO is synthesized in the microvasculature and regulates β-secretase activity which participate in APP processing (Austin et al., 2010). NOS2 also favors Aβ elimination by regulating MMP-9/TIMP-1 expression, which is a key enzyme that degrades amyloid (Ridnour et al., 2012). In addition, it has been shown that chronic NO formation may alter insulin-degrading enzyme activity (Kummer et al., 2012). NO synthesis during AD development could also contribute to NFT formation. In co-cultures of astrocytes and hippocampal neurons of rats, exposure to Aβ 25–35 was found to increase NO levels, which correlated with increased hyperphosphorylated Tau protein. Interestingly, the use of a NOS inhibitors reduced Tau phosphorylation levels (Saez et al., 2004).
http://journal.frontiersin.org/article/10.3389/fnint.2013.00059/full
Insulin Resistance and Alzheimer’s Disease
Alzheimer disease (AD) is known as a form of type III diabetes due to its similar cellular responses and pathogenesis. Insulin alters normal brain function and peripheral glucose metabolism, and conditions that are related to insulin dysregulation, such as obesity, diabetes mellitus, and cardiovascular disease, have potentially harmful effects on brain function. Many reports have demonstrated that insulin resistance increases age-related memory impairments and is a risk factor for AD. The molecular and cellular link between insulin resistance and AD, however, is unknown. We discuss the potential mechanisms of these metabolic disorders in the pathogenesis of AD. Glucose homeostasis is critical for energy maintenance, neurogenesis, neuronal survival, and synaptic plasticity, which are required for learning and memory. During insulin resistance, one develops reduced sensitivity to insulin, resulting in hyperinsulinemia, and this impairment in insulin signaling mediates the pathogenesis of AD, which manifests as brain inflammation, oxidative stress, alterations in amyloid beta (Aβ) levels, and cell death. Human and experimental animal studies have noted that drugs that modulate insulin resistance decrease the accumulation of Aβ in the brain and the cognitive impairments that are associated with AD. Therapeutic strategies that target the link between insulin resistance and AD might benefit the development of future AD drugs.
Insulin-like growth factors (IGF)
Insulin-like growth factors are polypeptides that are similar in sequence to proinsulin (Clemmons, 2007). Insulin-like growth factor 1, which stimulates cell growth and proliferation, especially in nerve cells, binds to IGF receptor and IRs (Jones et al., 2009). IGF-1 functions similarly to insulin receptor. IGF-1, a tyrosine kinase, initiates signaling cascades through IRS and enhances insulin activity (Clemmons, 2007).
Deficiency of or irresponsiveness to IGF-1 causes not only growth failure but also dyslipidemia (Twickler et al., 2003), insulin resistance (Conti et al., 2002; Twickler et al., 2003), and obesity (Rasmussen et al., 1995). According to recent longitudinal studies, these phenotypes are closely related to a high risk of neurodegenerative disorders, such as dementia and AD (Luchsinger et al., 2004; Ott et al., 1996; Ronnemaa et al., 2008). Similarly, mice in which IGF-1 has been deleted develop increased Aβ levels in the brain, suggesting that IGF-1 promotes the clearance of Aβ (Carro et al., 2002). Moreover, IGF-1 mediates transient site-selective increases in tau phosphorylation—hyperphosphorylated tau is a pathological hallmark of AD—in primary cortical neurons via GSK-3 pathways (Lesort and Johnson, 2000).

Schematic diagram of the effects of peripheral insulin resistance on insulin, insulin-degrading enzyme (IDE) and Aβ levels. Peripheral insulin resistance and hyperinsulinemia triggers an excess release of FFA from adipocytes to liver and muscles. A three-fold increase of FFAs reduce insulin-dependent skeletal muscle glucose uptake by 50% (Roden et al., 1996), thereby hampering further insulin signal transduction (Hotamisligil et al., 1996; Schinner et al., 2005). In addition, Increased peripheral FFA levels invoke elevations in TNFα in the periphery and in the CNS, which may result in increased accumulation of Aβ. Chronic elevations in plasma Aβ may result in increased transport of Aβ into the brain. In contrast, peripheral hyperinsulinemia decrease insulin transport into the CNS. Low insulin level in CNS, and low level of IDE may contribute to the formation of senile plaques via disability to degrade Aβ peptides (Jones et al., 2009), further promoting intraneuronal Aβ accumulation, which is the hallmark of AD. <modified from Craft’s paper (Craft, 2007 )>
Insulin-degrading enzyme (IDE)
Insulin-degrading enzyme is the chief enzyme that degrades excess insulin and other substrates, including Aβ, a peptide that is implicated in the pathogenesis of AD (Wang et al., 2010). IDE knockout mice experienced decreased Aβ degradation, hyperinsulinemia, and hyperglycemia (Farris et al., 2003). As insulin levels rise, IDE expression increases to prevent the chronic activation of insulin (Zhao et al., 2004). Reduced IDE activity might contribute to the formation of senile plaques through the inability to degrade Aβ peptides (Jones et al., 2009).
http://www.intechopen.com/books/topics-in-the-prevention-treatment-and-complications-of-type-2-diabetes/insulin-resistance-and-alzheimer-s-disease