Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders
Contents
Mitochondrial ROS Production and Defense
Estimates indicate that approximately 0.2–0.5% of the oxygen consumed by the mitochondrion is converted into ROS, a number that is tissue-dependent and varies with the redox state of the organelle (Chance et al., 1979). Heavy metals, ethanol, and oxidative stress are known to exacerbate ROS formation (Auger et al., 2013; Lemire et al., 2014). ROS leakage in the ETC occurs predominantly at the sites of complex I (NADH ubiquinone oxidoreductase) and complex III (ubiquinone cytochrome c oxidoreductase) in the form of superoxide (O•−2) (Miwa and Brand, 2005; Treberg et al., 2011). However, auxiliary sites of ROS production, such as pyruvate dehydrogenase (PDH), alpha-ketoglutarate dehydrogenase (KGDH), and succinate dehydrogenase (SDH), do add to the oxidative burden of this organelle (Murphy, 2012). Within the mitochondrial matrix, Mn-dependent superoxide dismutases (MnSOD) are known to catalyze the dismutation of O•−2 to hydrogen peroxide (H2O2) that can be readily detoxified by glutathione peroxidase (GPX) (Wallace et al., 2010). However, if the concentration of H2O2 is not controlled, it diffuses to the cytoplasm and participates in a series of reactions generating other reactive compounds, such as hydroxyl radical (•HO). The indiscriminate nature of •HO, which reacts with lipids, nucleic acids, and amino acids, renders it short lived but highly dangerous in biological systems (Imlay, 2013). Furthermore, the diffusion-limited reaction of O•−2 with the gaseous signaling compound nitric oxide (NO•) forms peroxynitrite (ONOO−) (Auger and Appanna, 2015). The generation of this oxidant and nitrating agent can have potentially toxic ramifications via the nitration of tyrosine residues and s-nitrosylation of cysteine moieties on vital proteins in the mitochondrion (Pacher et al., 2007). Moreover, exposure to bioavailable cationic metals such as Al and Zn can lead to an increase in ROS and displace Fe from the active site of some proteins, such as aconitase (ACN) (Lemire et al., 2008; Han et al., 2013). Free Fe poses a threat to cells due to its participation in Fenton chemistry, which further increases the concentration of detrimental •HO (Ahlqvist et al., 2015).
Nitro-oxidative stress occurs when the production of these radicals is in excess of the ability of the cell to limit their reactivity. Given that the majority of cellular ROS is generated in the mitochondrion, which houses high-priority targets of these moieties, this organelle requires an intricately-regulated antioxidant system (Song et al., 2013). The aforementioned MnSOD, in tandem with mitochondrial glutathione (mGSH) as well as glutaredoxin and thioredoxin systems are responsible for limiting the accumulation of O•−2 and H2O2. GSH, which is synthesized in the cytosol and imported into the mitochondrion, reduces peroxide concentrations with the assistance of GPX, or peroxiredoxin, subsequently producing oxidized glutathione (GSSG) (Mari et al., 2009). The latter is converted back into GSH via NADPH-dependent glutathione reductase, thus maintaining the GSH redox cycle. The pool of mGSH represents 10–15% of the total cellular GSH content (Ribas et al., 2014). GSH renewal is assured by a number of NADPH-generating enzymes in the mitochondrion, including malic enzyme (ME), NADP-dependent isocitrate dehydrogenase (ICDH), and a matrix-localized glucose-6-phosphate dehydrogenase (G6PDH) (Figure 2) (Mailloux and Harper, 2010; Yin et al., 2012a). In humans, the absence of various metabolic strategies aimed at neutralizing oxidative stress that are operative in other organisms gives rise to ineffective mitochondrial enzymes and disease can ensue (Chenier et al., 2008; Mailloux et al., 2011b).
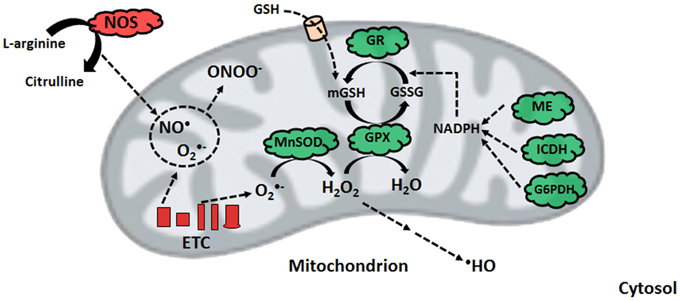
Nitro-oxidative stress production and detoxification. Within the mitochondrion, superoxide from electron transport chain activity can be readily converted to peroxide or react with nitric oxide to form peroxynitrite. To limit peroxide diffusion and synthesis of the hydroxyl radical, organisms maintain a pool of mitochondrial glutathione, whose renewal is orchestrated by glutathione reductase with the help of various NADPH-generating enzymes. ETC, electron transport chain; G6PDH, glucose-6-phosphate dehydrogenase; GPX, glutathione peroxidase; GR, glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; ICDH, NADP-dependent isocitrate dehydrogenase; ME, malic enzyme; MnSOD, manganese superoxide dismutase; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NOS, nitric oxide synthase.
Liver Mitochondria and Diabetes Pathogenesis
Diabetes mellitus is a multifactorial metabolic disorder characterized by insensitivity to insulin, a hormone regulating the absorption of glucose from blood to bodily tissues. The liver is a primary target of insulin and glucagon signaling, regulating glycogen synthesis, and gluconeogenesis in hepatocytes. It is a well-recognized fact that increased ROS and diminished mitochondrial ATP production are molecular hallmarks of type 2 diabetes mellitus (T2DM) (Szendroedi et al., 2012). Indeed, diabetic patients have higher levels of mitochondrial protein and lipid oxidation. An increase in mutations and low concentrations of mtDNA have also been reported. More specifically, an augmentation in the lipid peroxidation product 4-hydroxynonenal (4-HN) and DNA oxidation biomarker 8-hydroxy-2′-deoxyguanosine in addition to an up-regulated NADPH oxidase and decreased GSH:GSSG ratio are common symptoms of the disorder (Noriega-Cisneros et al., 2013). Lowered activity of complex I/IV as well as MnSOD are signs of T2DM progression, and likely the result of increased nitro-oxidative stress affecting key moieties such as Fe-S clusters, heme groups, and mtDNA coding for ETC components (Pagano et al., 2014). What remains unclear is whether these biomolecular events precede, accompany or are a consequence of the pathogenesis of diabetes. In Otsuka Long-Evans Tokushima fatty rats, a commonly studied model of obesity and T2DM, hepatic mitochondrial dysfunction occurs weeks in advance of the development of insulin resistance. Lower SOD and increased concentrations of GSSG were detectable at 5 weeks of age. Decreased activity of carnitine palmitoyltransferase I, the rate-limiting step of mitochondrial fatty acid entry, was accompanied by impaired fatty acid oxidation (Rector et al., 2010). Excess energy intake in the form of over-nutrition leads to an increase in circulating free fatty acids. As lipid infusion has been shown to reduce insulin sensitivity, defective lipid metabolism stemming from mitochondrial disorder appears to be a key initiator of insulin resistance and subsequently, T2DM. In the isolated liver, gluconeogenesis, the generation of glucose, is dependent on mitochondrial fat oxidation. It is noteworthy that while β-oxidation and ketogenesis are dysfunctional in the diabetic liver, pyruvate carboxylase, and TCA cycle flux appear to be elevated. This is in contrast to the mitochondria from skeletal muscle of diabetic rat models, where TCA cycle activity appears to be down-regulated. As such, the build-up of NADH and fatty acid concentrations in conjunction with the lowered activity of ETC complexes may further compound the ROS burden, thus bringing about insulin-resistance in the liver of mammalian organisms (Kim et al., 2008). Hence, mitochondrial dysfunction coupled with diminished ATP synthesis is an important contributor to diabetes.
Therapeutic Cues
Despite the variance in phenotype stemming from the plethora of liver disorders, there exists a significant overlap in the biomolecular pathogenesis at the mitochondrial level. Defective oxidative phosphorylation, increased ROS and RNS, diminished β-oxidation, and enhanced lipogenesis are some of the recurring commonalities across the spectrum of hepatic diseases. Therefore, mitigating the oxidative burden and bolstering the mitochondrion’s capacity to produce ATP and break down fatty acids can have significant health benefits. The interplay between mitochondrial function and mitochondrial biogenesis suggests that increasing the number of this organelle can alleviate the symptoms of mitochondrial disorders. Indeed, treatment with thiazolidinediones, synthetic peroxisome proliferator-activated receptor gamma (PPAR-γ) ligands, has been reported to improve insulin resistance partially through increased mitochondrial biogenesis (Bogacka et al., 2005). The antidiabetic drug metformin has also been reported to increase mitochondrial numbers through activation of PGC-1α and adenosine monophosphate kinase (AMPK) signaling, while also lowering ROS concentrations (Kim et al., 2008).
Mitochondria-targeted antioxidants can also be exploited for the remediation of liver disorders. For instance, the beneficial effects of vitamin E and C supplementation on tempering ROS levels are well described (Nassir and Ibdah, 2014b). The ONOO− scavenger metalloporphyrin has positive effects on mitochondrial redox state, as demonstrated by restored activity of ALDH2 and ATP synthase (Moon et al., 2008). Treatment with resveratrol, a polyphenolic antioxidant found in grapes, can prevent high fat diet-induced steatosis through regulation of metabolic regulators of energy such as SIRT1 and AMPK (Poulsen et al., 2012).
Keto acid supplementation in the form of KG or pyruvate can relieve nitro-oxidative stress directly via their antioxidant properties while restoring other mitochondrial functionalities indirectly. For example, KG treatment in HepG2 cells increases levels of L-carnitine and restores mitochondrial β-oxidation (Lemire et al., 2011).
Pyruvate supplementation drives the pyruvate carboxylase and ME reactions, thus augmenting levels of NADPH and mitochondrial citrate. The latter effluxes to the cytosol and suppresses phosphofructokinase activity, diverting glucose to the production of NADPH via the pentose phosphate pathway. The end result of this metabolic reconfiguration is an increased GSH:GSSG ratio, a parameter leading to reduced oxidative damage. Indeed, pyruvate supplementation suppresses mtDNA damage and protein degradation (Tanaka et al., 2007; Liu et al., 2011).
While supplements may slow the progression of liver disease, preventative measures such as reduced caloric intake, exercise, and lower alcohol consumption are key factors in restoring mitochondrial ability. Exercise and physical activity has been shown to improve mitochondrial function and insulin sensitivity in T2DM patients and subjects with NAFLD. Aerobic exercise may stimulate mitochondrial biogenesis through increased expression of PGC-1α (Kim et al., 2008). Furthermore, endurance exercise and caloric restriction increase mitochondrial size, number, and oxidative phosphorylation, thus improving cellular bioenergetics. Additionally, dietary choices such as increased consumption of polyunsaturated fatty acids and antioxidant-containing foods can prevent protein oxidation and mitochondrial dysfunction (Nassir and Ibdah, 2014b). These lifestyle decisions ultimately lead to higher oxidative ATP production, increased fatty acid oxidation and lower hepatic fat accumulation, thus limiting the pathogenesis of liver disorders.
Conclusion
The pathogenesis of liver disease is a complex and multifactorial process where environmental and genetic signals converge upon the organ, causing failure to accomplish its vast biological functions. While there exists several disparities in the means by which liver disorders progress and present themselves, mitochondrial dysfunction is a common link in most hepatic diseases. The role of this organelle in the aerobic synthesis of ATP, oxidation of fatty acids and apoptotic signaling renders it indispensable to cellular function, particularly in energy-intensive organs like the liver. As such, injury to mitochondria which give way to dysfunction generally precedes the development of such disorders as T2DM, NAFLD, ALD, and HCC. Increased nitro-oxidative stress results in lipid peroxidation and protein oxidation and consequently, inactivation of ETC complexes and mtDNA mutations or deletions. Therapeutic and pharmacological strategies to decrease the oxidative burden and maintain a healthy mitochondrial redox state can halt or reverse the progression of various liver diseases, particularly when combined with healthy lifestyle habits (Figure 5). As such, mitochondria-targeted treatments which bolster the ability of this organelle to execute its functions can be implemented to alleviate liver diseases and thus should be explored more rigorously for their therapeutic potential.
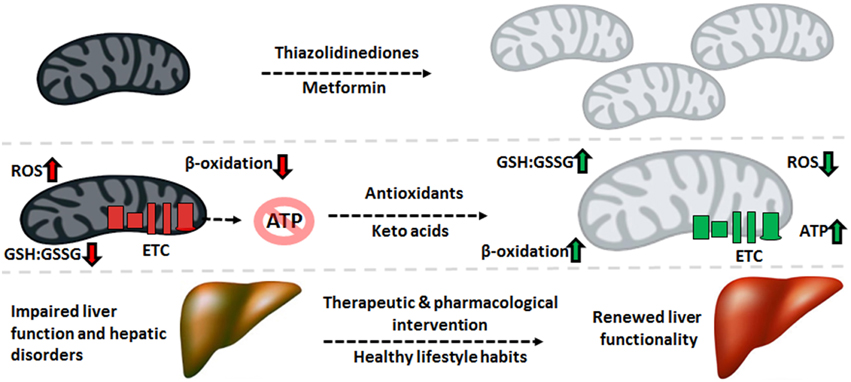
Therapeutic cues aimed at mitochondrial restoration. Pharmaceutical compounds which increase mitochondrial biogenesis and mitochondria-targeted antioxidants geared to diminishing the nitro-oxidative burden can be applied to reverse the molecular events underlying the pathogenesis of liver disorders, renewing the function of this crucial organ. ATP, adenosine triphosphate; ETC, electron transport chain; GSH, reduced glutathione; GSSG, oxidized glutathione; ROS, reactive oxygen species.
http://journal.frontiersin.org/article/10.3389/fcell.2015.00040/full
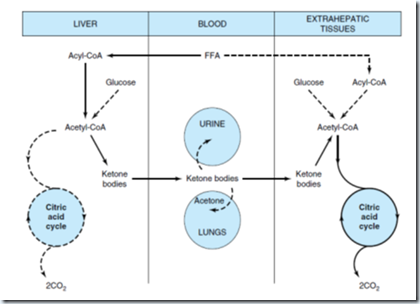
Formation, utilization, and excretion of ketone bodies.
Solid arrow indicates the main pathway
.