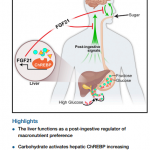
Branched-chain amino acids in metabolic signalling and insulin resistance
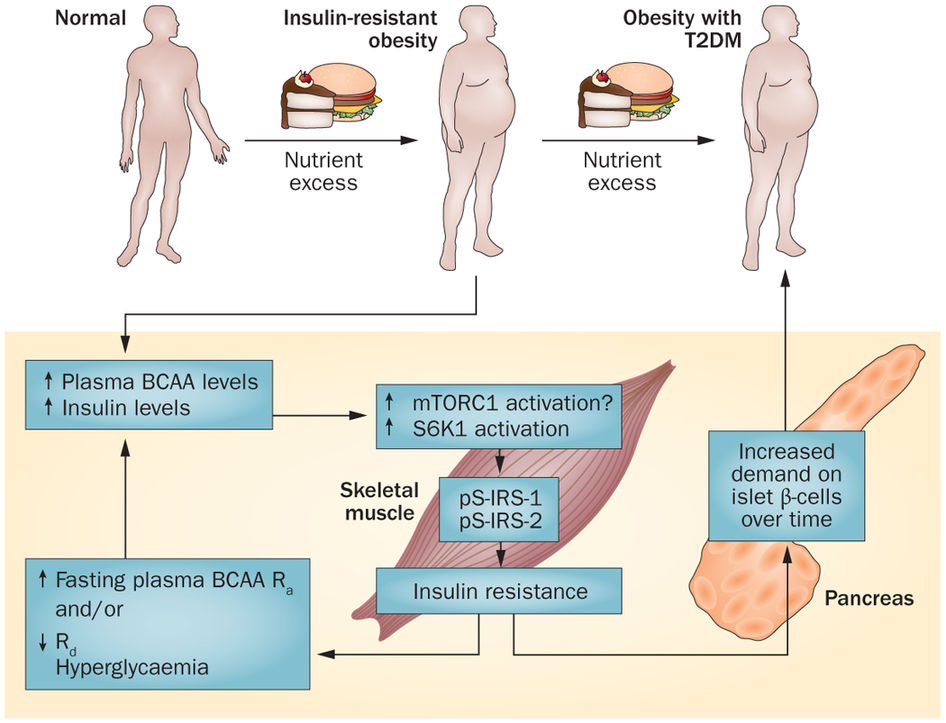
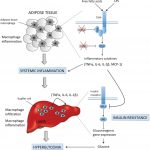
Branched-chain amino acids (BCAAs) are important nutrient signals that have direct and indirect effects. Frequently, BCAAs have been reported to mediate antiobesity effects, especially in rodent models. However, circulating levels of BCAAs tend to be increased in individuals with obesity and are associated with worse metabolic health and future insulin resistance or type 2 diabetes mellitus (T2DM). A hypothesized mechanism linking increased levels of BCAAs and T2DM involves leucine-mediated activation of the mammalian target of rapamycin complex 1 (mTORC1), which results in uncoupling of insulin signalling at an early stage. A BCAA dysmetabolism model proposes that the accumulation of mitotoxic metabolites (and not BCAAs per se) promotes β-cell mitochondrial dysfunction, stress signalling and apoptosis associated with T2DM. Alternatively, insulin resistance might promote aminoacidaemia by increasing the protein degradation that insulin normally suppresses, and/or by eliciting an impairment of efficient BCAA oxidative metabolism in some tissues. Whether and how impaired BCAA metabolism might occur in obesity is discussed in this Review. Research on the role of individual and model-dependent differences in BCAA metabolism is needed, as several genes (BCKDHA, PPM1K, IVD and KLF15) have been designated as candidate genes for obesity and/or T2DM in humans, and distinct phenotypes of tissue-specific branched chain ketoacid dehydrogenase complex activity have been detected in animal models of obesity and T2DM.
https://www.nature.com/articles/nrendo.2014.171?message-global=remove
Contents
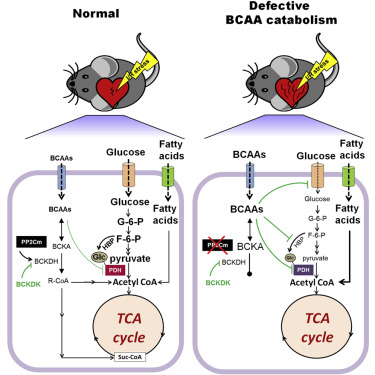
Defective Branched-Chain Amino Acid Catabolism Disrupts Glucose Metabolism and Sensitizes the Heart to Ischemia-Reperfusion Injury
Highlights
•Branched-chain amino acid (BCAA) catabolism regulates glucose metabolism in the heart
•High levels of BCAAs selectively disrupt mitochondrial pyruvate utilization
•Downregulation of HBP by chronic accumulation of BCAAs inactivates PDH
•Defective BCAA catabolism sensitizes the heart to ischemia-reperfusion insult
Summary
Elevated levels of branched-chain amino acids (BCAAs) have recently been implicated in the development of cardiovascular and metabolic diseases, but the molecular mechanisms are unknown. In a mouse model of impaired BCAA catabolism (knockout [KO]), we found that chronic accumulation of BCAAs suppressed glucose metabolism and sensitized the heart to ischemic injury. High levels of BCAAs selectively disrupted mitochondrial pyruvate utilization through inhibition of pyruvate dehydrogenase complex (PDH) activity. Furthermore, downregulation of the hexosamine biosynthetic pathway in KO hearts decreased protein O-linked N-acetylglucosamine (O-GlcNAc) modification and inactivated PDH, resulting in significant decreases in glucose oxidation. Although the metabolic remodeling in KO did not affect baseline cardiac energetics or function, it rendered the heart vulnerable to ischemia-reperfusion injury. Promoting BCAA catabolism or normalizing glucose utilization by overexpressing GLUT1 in the KO heart rescued the metabolic and functional outcome. These observations revealed a novel role of BCAA catabolism in regulating cardiac metabolism and stress response.
http://www.sciencedirect.com/science/article/pii/S1550413116305836

Protein and amino acid restriction, aging and disease: from yeast to humans
Highlights
•Protein or AA restriction has been shown to be as potent as calorie restriction in extending healthspan in multiple model organisms.
•AA restriction affects lifespan partly through modulation of the amino acid sensing pathways TOR and GCN2.
•Human epidemiological studies highlight the detrimental effects of high protein diets, in particular animal-derived protein sources in contrast to plant-based sources.
•Epidemiological studies indicate that low protein diets are associated with lower risk of chronic and age-related diseases such as CVDs, diabetes, and cancer.
Many of the effects of dietary restriction (DR) on longevity and health span in model organisms have been linked to reduced protein and amino acid (AA) intake and the stimulation of specific nutrient signaling pathways. Studies in yeast have shown that addition of serine, threonine, and valine in media promotes cellular sensitization and aging by activating different but connected pathways. Protein or essential AA restriction extends both lifespan and healthspan in rodent models. In humans, protein restriction (PR) has been associated with reduced cancer, diabetes, and overall mortality. Thus, interventions aimed at lowering the intake of proteins or specific AAs can be beneficial and have the potential to be widely adopted and effective in optimizing healthspan.
http://www.cell.com/trends/endocrinology-metabolism/fulltext/S1043-2760(14)00127-1
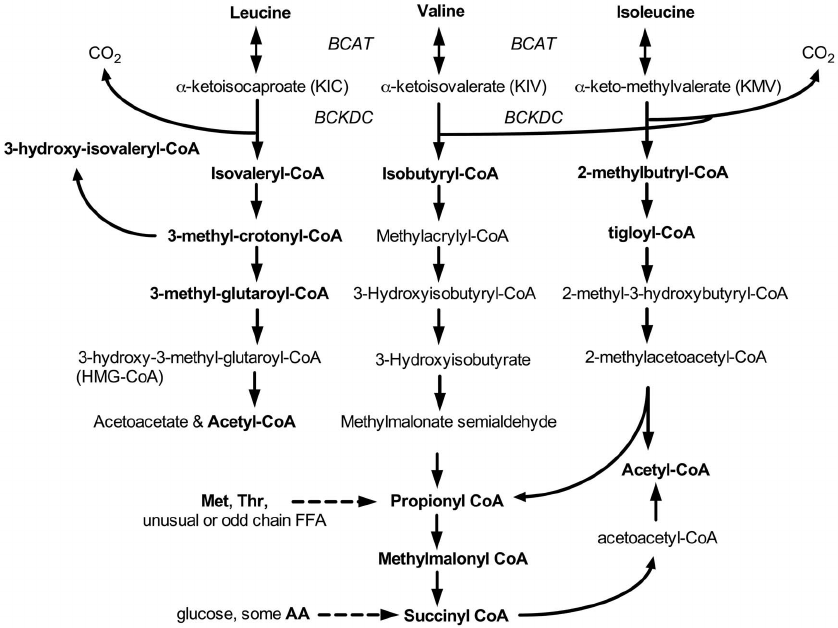
Role of Food Intake and BCAA Oxidation in Elevating BCAAs in Obesity.
https://www.researchgate.net/publication/236079012_Leucine_and_Protein_Metabolism_in_Obese_Zucker_Rats