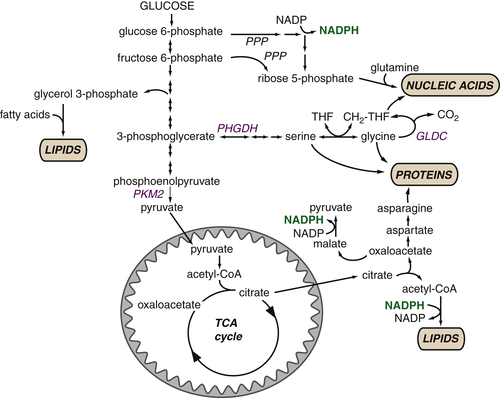
Glucose metabolism provides critical intermediates for cell growth Glycolytic intermediates provide the substrates for the pentose phosphate pathway (PPP) that generates the ribose 5-phosphate that is critical for nucleotide biosynthesis. The oxidative arm of the PPP, which utilizes glucose 6-phosphate, additionally generates NADPH. Glycerol 3-phosphate, which contributes the glycerol head groups for phospholipid biosynthesis, is formed from the intermediate dihydroxyacetone phosphate. 3-Phosphoglycerate provides the foundation for the serine synthesis pathway, which can further fuel glycine production for protein synthesis. This pathway also contributes to the pool of one-carbon units (CH2-THF) that are used in nucleotide biosynthesis. Glucose-derived citrate provides the acetyl-CoA that represents the fundamental building block for the synthesis of the fatty acids that comprise cellular lipids. Key metabolic enzymes are shown in blue; NADPH required for biosynthetic reactions is shown in green. CH 2 -THF, methylene tetrahydrofolate; GLDC, glycine decarboxylase; PHGDH, phosphoglycerate dehydrogenase; PKM2,pyruvate kinase M2 isoform; PPP, pentose phosphate pathway; THF, tetrahydrofolate.
Glycolysis Provides Key Intermediates to Support Cell Growth
The preceding examples provide clues for how rapid aerobic glycolysis might provide a growth or survival advantage to tumor cells. However, we now know that aerobic glycolysis as Warburg described it—the conversion of glucose to lactate—is not the exclusive fate of glucose in cancer cells. Glucose is a major source of many of the building blocks of macromolecular biosynthesis, and it is clear that these various anabolic fates of glucose are an important component of the elevated glycolysis in cancer cells.
Glucose-Derived Metabolites Contribute to All Classes of Macromolecules
Glycolytic flux provides substrates for biosynthetic reactions that are required for the synthesis of lipids, nucleotides, and proteins (
Figure 13-5 ). Glycolytic intermediates can be diverted to the pentose phosphate pathway to produce ribose 5-phosphate, which serves as the foundation for the de novo synthesis of purines and pyrimidines. The oxidative arm of the pentose phosphate pathway also generates NADPH, which can provide reducing equivalents for nucleotide and fatty acid synthesis. Similarly, the glycerol required for phospholipid synthesis is derived from the glycolytic intermediate dihydroxyacetone phosphate. As the major component of mammalian cell membranes, phospholipid synthesis is critical for cell growth. Glycolytic intermediates can also contribute to amino acid synthesis: 3-Phosphoglycerate can be converted to serine and ultimately glycine.
A series of recent studies documenting tumors whose growth depends on diversion of glycolytic flux to anabolic pathways lends support to the hypothesis that glycolytic intermediates play a key role fueling tumor growth. Phosphoglycerate dehydrogenase (PHGDH), the enzyme that catalyzes the first step of the serine synthesis pathway that branches from glycolysis, is frequently amplified in melanoma and breast cancers, and flux into the serine synthesis pathway can support tumor cell growth.
24,25 Similarly, multiple tumor types display high levels of glycine decarboxylase (GLDC), which generates methylene tetrahydrofolate necessary for pyrimidine synthesis, and GLDC expression can promote cellular transformation and tumorigenesis.
26
Macromolecular Synthesis, Not ATP, May Be the Major Purpose of Elevated Glycolysis in Cancer Cells
The importance of glycolytic intermediates as biosynthetic building blocks supporting cell proliferation was underscored by the observation that most proliferating cells express the M2 isoform of pyruvate kinase (PK), whereas quiescent cells express the PKM1 isoform.
27 Pyruvate kinase catalyzes the final ATP-producing step of glycolysis, converting phosphoenolpyruvate to pyruvate. Perhaps counterintuitively, PKM2 has less intrinsic activity than PKM1; PKM2 is also inhibited by growth factor signaling. These observations suggest that
inhibiting the final step of glycolysis can be beneficial for cell growth. By slowing the final step of glycolysis, PKM2 enables accumulation of upstream glycolytic intermediates that can then be diverted into biosynthetic pathways. These findings indicate that proliferating cells do not undergo glycolysis for the sole purpose of ATP production; rather, they appear to selectively slow pyruvate production in order to maximize the flow of glucose carbons into anabolic pathways.
Expression of PKM2 may have the added benefit of reducing ATP production. Although ATP is required for anabolic reactions, NADPH is quantitatively more important. Biosynthesis of many macromolecules requires more reducing equivalents than energy from ATP.
1 Consequently, if all glucose were oxidized completely, ATP production would far outstrip NADPH production, thus greatly circumscribing potential cell growth. Furthermore, ATP is a potent inhibitor of phosphofructokinase-1, a key enzyme in the regulation of glycolysis, and so ATP produced in excess of biosynthetic demands would have the adverse effect of blocking glycolysis, further reducing NADPH production.
Although cancer cells preferentially reduce pyruvate kinase activity, the final step in glycolysis cannot be abrogated completely, as pyruvate fulfills many important metabolic roles. The pyruvate produced at this step has three fates: It can be transaminated to alanine, reduced to lactate to regenerate NAD
+, or imported to the mitochondrion, where it is converted to acetyl-CoA that can enter the TCA cycle. In many tumors, glucose consumption is high enough to support anabolic pathways while also maintaining pyruvate flux into the mitochondria and still resulting in high amounts of lactate secretion. It is likely that reducing pyruvate kinase activity slows glucose flux through glycolysis in order to increase the likelihood that glucose can be diverted into anabolic pathways rather than being completely and rapidly exported as lactate or oxidized in the mitochondria. Given the high rates of lactate produced from glucose (as much as 90% of glucose is converted to lactate and alanine in glioblastoma cells),
28 cancer cells still have appreciable pyruvate consumption. It is likely that despite the high percentage of pyruvate that is secreted as lactate, the rate of glycolysis is so high that sufficient pyruvate remains to fuel the TCA cycle.
The TCA Cycle as a Biosynthetic Hub
Just as glycolytic intermediates are harnessed to support anabolic pathways, proliferating cells rewire the TCA cycle to prioritize cell growth over ATP generation. In its traditional form, the TCA cycle begins with the condensation of pyruvate-derived acetyl-CoA with oxaloacetate to form citrate (
Figure 13-6 ). A series of reactions in the mitochondrial matrix convert citrate back to oxaloacetate, releasing two carbons as two molecules of carbon dioxide and producing three molecules of NADH and one molecule of FADH
2. NAD
+ and FAD are regenerated when NADH and FADH
2 donate their electrons to support oxidative phosphorylation. The cycle can continue by combining the recycled oxaloacetate with a new molecule of acetyl-CoA. In this manner, the oxidation of acetyl-CoA is used to maximize ATP production.
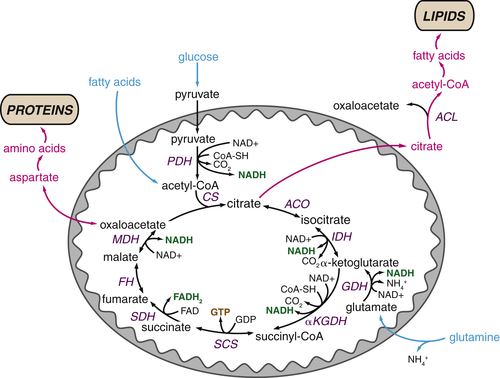
FIGURE 13-6 The tricarboxylic acid (TCA) cycle supports macromolecular biosynthesis Pyruvate derived from glucose enters the mitochondrion and is converted to acetyl-CoA by pyruvate dehydrogenase (PDH), and fatty acids are oxidized to produce acetyl-CoA units within the mitochondrion. Acetyl-CoA condenses with oxaloacetate to form citrate, which proceeds through the TCA cycle in highly oxidative tissues, releasing two carbons as carbon dioxide and regenerating oxaloacetate for further cycling. These reactions maximize ATP production from acetyl-CoA by reducing the electron carriers NADH and FADH2 to fuel the electron transport chain. In proliferating cells, citrate is also exported from the mitochondrion to the cytosol, where it can provide the acetyl-CoA required for the synthesis of fatty acids and cholesterol. Similarly, oxaloacetate can form aspartate and other amino acids, contributing to intracellular amino acid pools for protein synthesis. Both of these reactions deplete TCA cycle intermediates; consequently, cells must replenish TCA cycle metabolites in order to regenerate the oxaloacetate necessary for citrate synthesis. Most cells use glutamine to fulfill this anaplerotic role: glutamine is converted to glutamate and then α-ketoglutarate, preserving TCA cycle flux. Metabolites that fuel the TCA cycle are shown in light blue. Metabolite efflux from the TCA cycle is shown in pink. Enzymes are shown indark blue, reduced electron carriers in green, and high-energy triphosphates in orange. PDH, pyruvate dehydrogenase; ACL, ATP-citrate lyase;ACO, aconitase; CoA-SH, coenzyme A; CS, citrate synthase; FH, fumarate hydratase; GDH, glutamine dehydrogenase; IDH, isocitrate dehydrogenase; αKGDH, α-ketoglutarate dehydrogenase; MDH, malate dehydrogenase; SCS, succinyl-CoA synthetase; SDH, succinate dehydrogenase.
TCA Cycle Metabolites Are Required for Macromolecular Biosynthesis
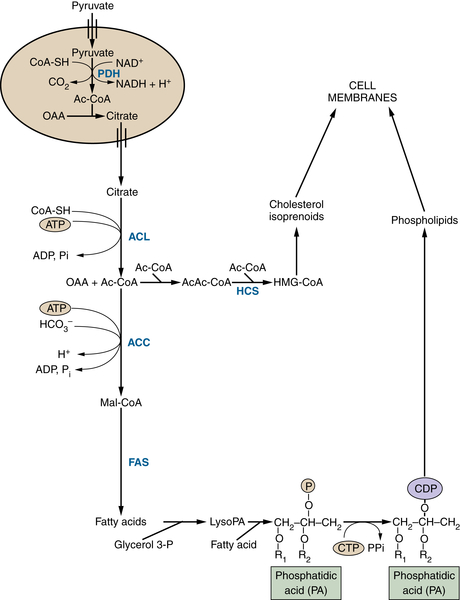
In proliferating cells, citrate is not only oxidized in the TCA cycle but also can be exported to the cytosol where it is converted back to oxaloacetate and acetyl-CoA by the
enzyme ATP citrate lyase (ACL). Cytosolic acetyl-CoA provides the substrate for the synthesis of fatty acids, cholesterol, and prostaglandins (
Figure 13-7 ). Through this pathway, glucose acts as the major substrate for de novo lipogenesis; consequently,
ACL inhibition blocks cell proliferation and inhibits tumor growth.
28–30Citrate is not the only TCA cycle metabolite that has an important biosynthetic role. Oxaloacetate and α-ketoglutarate can provide the carbon backbone for nonessential amino acids, which are used for protein and nucleic acid synthesis (see
Figure 13-6). In this manner, multiple TCA cycle metabolites are diverted to other pathways that support cell growth.
Glutamine Plays Several Roles Supporting the TCA Cycle and Anabolic Metabolism
Although efflux of TCA cycle metabolites supports anabolic reactions, this results in depletion of TCA cycle intermediates from the mitochondrial matrix. Growing cells must draw on alternative sources to maintain the oxaloacetate required for citrate synthesis. Replenishment of TCA cycle intermediates, a process known as
anaplerosis, is thus critical to maintain both a constant supply of metabolites for synthesis and appropriate ATP production through oxidative phosphorylation.
Most proliferating cancer cells meet their anaplerotic needs through the catabolism of glutamine (see
Figure 13-6). Glutamine is converted to the TCA cycle intermediate α-ketoglutarate through a metabolic pathway known as glutaminolysis. Tumor cells take up high levels of glutamine, often in great excess of any other amino acid, and studies using carbon labeling techniques have demonstrated that glutamine is the major source of carbon for the cellular oxaloacetate pool in cancer cell lines.
28
FIGURE 13-7 Glucose-derived acetyl-CoA fuels lipid and sterol synthesis Pyruvate derived from glycolysis can generate citrate in the mitochondrion. This citrate can be exported to the cytosol to provide acetyl-CoA for the synthesis of fatty acids, phospholipids, cholesterol, and isoprenoids, all of which are critical for membrane biogenesis and function. Metabolites are in black; enzymes are in blue. ACC, Acetyl-CoA carboxylase; Ac-CoA, acetyl-CoA; ACL, ATP-citrate lyase; CDP, cytidine diphosphate; CoA-SH, coenzyme A; CTP, cytidine triphosphate; FAS, fatty acid synthetase; glycerol-3-P, glycerol 3-phosphate; HCS, HMG-CoA synthetase; HMG-CoA, 3-hydroxy-3-methylglutaryl CoA; lyso PA,lysophosphatidic acid; Mal-CoA, malonyl-CoA; OAA, oxaloacetate; Pi, inorganic phosphate; R, acyl group on a lipid molecule.
By maintaining flux through the TCA cycle—and therefore delivery of electrons to the electron transport chain—glutamine plays a critical role maintaining mitochondrial bioenergetics in many cancer cells. Consequently, many cancer cell lines are absolutely dependent on glutamine for survival. Addition of cell-permeable TCA cycle analogs can rescue death of glutamine-deprived cells, highlighting the importance of glutamine as an anaplerotic substrate in cancer cells.
31,32Proliferating cells rely on glutamine to fulfill additional biosynthetic roles.
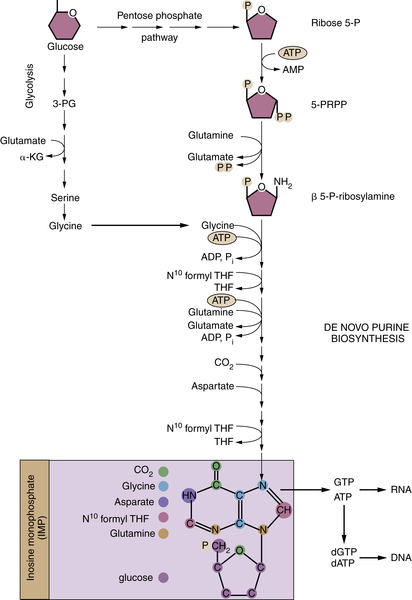
First, glutamine provides an important source of nitrogen for synthesis of nonessential amino acids and nucleotides. Glutamine is required for two independent steps in purine nucleotide synthesis, and oxaloacetate-derived aspartate is required for a third (
Figure 13-8 ). Similarly, two steps of pyrimidine synthesis require glutamine. In all cases, glutamine donates nitrogen in the form of an amide group and is converted to glutamic acid, which provides a major source of nitrogen for amino acid synthesis. Transaminases can transfer the amine group from glutamic acid to α-ketoacids, which are themselves derived from the catabolism of glucose or glutamine, producing alanine, serine, aspartate, and ornithine. In turn, these amino acids act as precursors for the synthesis of glycine, cysteine, arginine, and asparagine. Likewise, two enzymatic reactions directly convert glutamate to proline. In this manner,
glucose and glutamine contribute to the synthesis of every nonessential amino acid except for tyrosine, which is directly produced from the essential amino acid phenylalanine.
FIGURE 13-8 Nucleotide biosynthesis requires multiple metabolic inputs Nucleotide biosynthesis requires inputs from several metabolic pathways, highlighting why cells must coordinately regulate multiple metabolic pathways during proliferation. The de novo synthesis of purines (shown) and pyrimidines requires glucose, several amino acids, and one-carbon groups from folate metabolism. The origin of individual carbons and nitrogens on inosine monophosphate (IMP), the precursor to GTP and ATP, are color-coded in the purple box. α-KG, α-ketoglutarate; N10formyl THF, N10 formyl tetrahydrofolate; 3-PG, 3-phosphoglycerate; β 5-P-ribosylamine, β 5-phosphate-ribosylamine; PRPP, 5-phosphoribosyl pyrophosphate; ribose 5-P, ribose 5-phosphate; P, phosphate group.
Intriguingly, cancer cells may convert up to 60% of glutamine carbon into lactic acid, an ostensibly wasteful secretion analogous to the Warburg effect.
28 One possible explanation for this behavior is that the conversion of glutamine to lactate produces NADPH, which is absolutely required for many anabolic reactions. NADPH is produced when malic enzyme converts glutamine-derived malate to pyruvate, which is subsequently reduced to lactate and secreted. In this manner, glutamine may contribute to all three needs of proliferating cells: Glutamine can provide the carbon and nitrogen for most metabolic building blocks, maintain ATP production to support bioenergetics, and generate reducing equivalents required for many anabolic reactions.
TCA Cycle Rearrangements Highlight the Role of TCA Cycle Metabolites as Biosynthetic Precursors
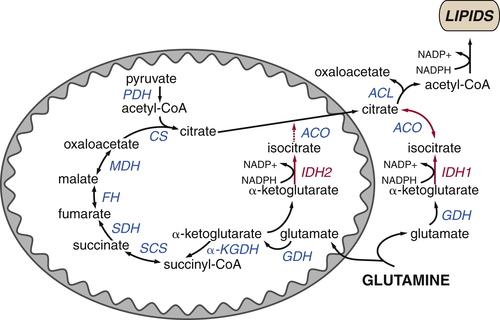
In proliferating cancer cells, the TCA cycle can behave more as a source of anabolic substrates rather than a bona fide cycle. A prime example of how the TCA cycle prioritizes biosynthetic reactions is the reductive carboxylation of α-ketoglutarate to isocitrate. During conditions of hypoxia or mitochondrial dysfunction, NADH accumulates from a combination of increased glycolysis and reduced oxidation of NADH by the electron transport chain. Depletion of oxidized NAD+ poses a bioenergetic challenge for the cell: Both the conversion of glucose-derived pyruvate to acetyl-CoA and the production of oxaloacetate through the enzymes of the TCA cycle require NAD+. Furthermore, during periods of acute hypoxia, pyruvate is converted to lactate at the expense of acetyl-CoA. Both of these events would strongly impair citrate production and thus cell growth. How, then, do hypoxic cells proliferate?
Several groups have demonstrated that under these conditions, the TCA cycle can partially function in reverse (for examples, see Refs.
33–
35). Normally, isocitrate dehydrogenase (IDH) catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate, consuming NAD
+ and producing NADH. When NAD
+ is limiting, both the mitochondrial and cytosolic isoforms of IDH can function in reverse, carboxylating α-ketoglutarate to isocitrate and producing oxidized NADP
+ (
Figure 13-9 ). In this manner, cells can use glutamine-derived α-ketoglutarate to generate citrate, providing a mechanism to maintain anabolic reactions even under hypoxic conditions that characterize the tumor microenvironment.
Genetic Mechanisms Driving Cancer Cell Metabolism
Metabolic transformation is a common feature of tumorigenesis, occurring in many tumor types in diverse tissues. It is logical that metabolic transformation and oncogenic transformation would go hand in hand: As cancer cells acquire mutations that enable pathological proliferation, they must also acquire the means to support cell growth. Thus, many of the mutations that promote cancer growth also trigger metabolic reprogramming. This section discusses several common genetic oncogenic events that reprogram metabolism to support tumor growth.
FIGURE 13-9 Reductive carboxylation supports lipid synthesis during hypoxia Citrate levels can be maintained even in cells with defective TCA cycles or during periods of hypoxia by the reductive metabolism of glutamine-derived α-ketoglutarate to citrate. Both mitochondrial and cytosolic isoforms of isocitrate dehydrogenase (IDH2 and IDH1, respectively) can function in reverse, reducing α-ketoglutarate to isocitrate and oxidizing NADPH to NADP+. Isocitrate is reversibly converted to citrate, which can be used to generate acetyl-CoA for lipid synthesis in the cytosol.
Activation of the PI3K Pathway Promotes Metabolic Transformation
In normal cells, extracellular cues regulate the intracellular signaling pathways that tightly coordinate metabolism and proliferation. In many cells, this coordination is accomplished by the phosphatidylinositol-3-kinase (PI3K) pathway, a highly conserved signaling pathway that responds to a variety of extracellular cues. When growth factors bind to their cell surface receptors, receptor tyrosine kinases (RTKs) activate PI3K, which phosphorylates phosphatidylinositol lipids at the plasma membrane. These phosphorylated lipids recruit and activate downstream effectors of the PI3K pathway, triggering an intracellular signaling cascade that promotes cell growth, survival, and metabolism.
Constitutive activation of PI3K provides a growth stimulus and is therefore a major mechanism of tumorigenesis. In normal cells, PI3K activity is tightly guarded by a number of factors, ensuring that pathway activity is set at appropriate levels. RTK activity is strictly controlled by growth factor availability. In addition, negative feedback loops prevent prolonged pathway activation: The lipid phosphatase PTEN dephosphorylates phosphatidylinositol species to dampen PI3K signaling.
Cancers exhibit a variety of mutations to circumvent this regulation and gain cell-autonomous activation of the PI3K pathway.
36 Amplifications or mutations of growth factor receptors occur in a variety of cancers. Common examples are the amplification of the Her2/Neu receptor in breast cancer and epithelial growth factor receptor (EGFR) in lung cancer. Similarly, deletion, downregulation, or loss of function of PTEN increases cancer susceptibility and promotes tumor progression. Several tumor types display activating mutations in the catalytic PI3K subunit, PIK3CA; likewise, amplification of PIK3CA and the major PI3K effector, Akt, are common oncogenic events. All together, genetic activation of the PI3K pathway is one of the most prevalent classes of tumorigenic mutations.
The metabolic network controls its own balance, chromatin structure and the biosynthesis of molecular cofactors; moreover, metabolic shifts are crucial in the response to oxidative stress and play a regulatory role in cancer.
(Visited 1,633 times, 1 visits today)