Beta cell dysfunction and insulin resistance
Contents
Beta Cell Physiology and Integrity
Adequate and proper beta cell function requires normal beta cell integrity which is critical for the appropriate response to perpetual fluctuating metabolic demand for insulin. Genes implicated in cell-cycle regulation are suggested to influence beta cell mass during development (Ashcroft and Rorsman, 2012). A decrease in beta cell mass of ≤60% has been reported in type 2 diabetes (Butler et al., 2003), which parallels the extent of reduction in glucose-stimulated insulin secretion (GSIS) (Del Guerra et al., 2005) but, however, considerably lower decrements have been found (Rahier et al., 2008). Although beta cell mass plays a role in type 2 diabetes, beta cell function rather than number is more critical in the etiology of type 2 diabetes (Ashcroft and Rorsman, 2012). Beta cells are resilient and will compensate to cope with insulin demand despite reduced numbers.
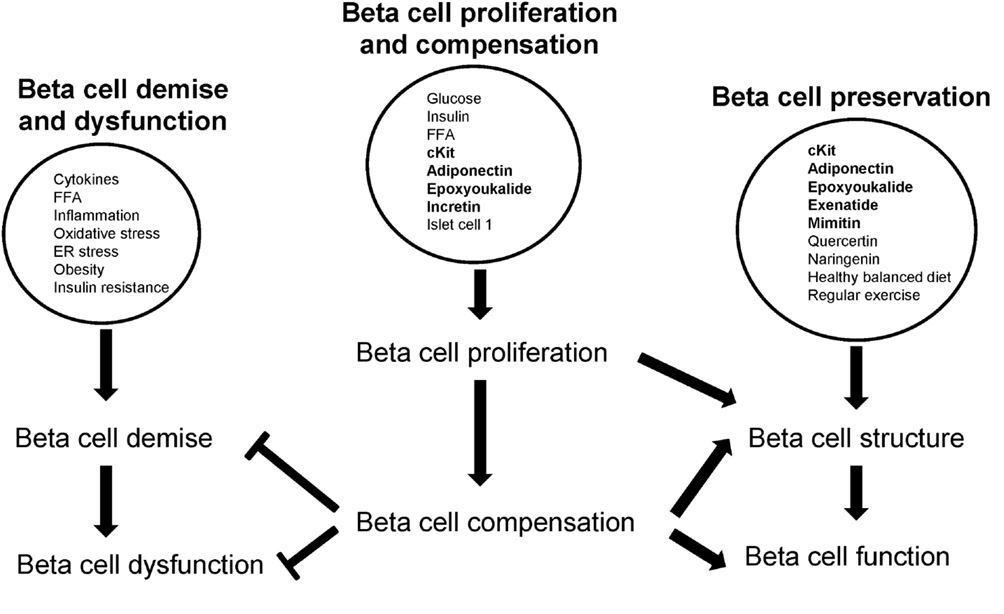
Beta Cell Demise and Dysfunction
Beta cell insults include cytokine-induced inflammation, obesity and insulin resistance, and overconsumption of saturated fat and free fatty acids (FFA). A progressive decline of beta cell function leading to beta cell exhaustion precedes beta cell demise (Ferrannini, 2010; Talchai et al., 2012). Loss of beta cell mass and function are central to the development of both type 1 and 2 diabetes (Stoffers, 2004).
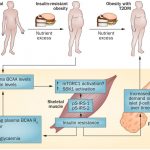
Obesity and Insulin Resistance
Obesity is a state of low grade inflammation. Adipose tissue is a major source of inflammation with the infiltration of macrophages as the primary source of cytokines in obese individuals (Stienstra et al., 2007). The beta cells secrete insulin to facilitate glucose uptake into glucose recipient organs (mainly the muscle, brain, liver, and adipose tissue). Obesity is a key risk factor for type 2 diabetes as it desensitizes glucose recipient organs to the action of insulin. Saturated fats are strongly associated with insulin resistance and beta cell dysfunction. High saturated fats in circulation, derived mainly from diets or even from lipolysis of fat depots, lead to fatty acids and glucose competing for uptake and metabolism in tissues. With persistent hyperglycemia, increased saturated FFA induce a glucolipotoxic state that is detrimental to beta cells by increasing oxidative stress, subsequently reducing insulin synthesis and secretion thereby compromising both beta cell structure and function.
The genetic architecture of quantitative indices of beta cell function and of insulin resistance differ markedly: given the same individuals, sample sizes, and biochemical measurements, more signals were described for beta cell function compared to insulin resistance (Dupuis et al., 2010; Voight et al., 2010). This infers that beta cell function supersedes insulin resistance as the critical determinant of type 2 diabetes. Further, as obesity is a critical determinant of insulin resistance, adiposity may modulate the genetic determinants of insulin resistance and contribute to the heterogeneity of type 2 diabetes (Manning et al., 2012). In addition, the adipokine hormones and proinflammatory cytokines that are produced by adipose tissue can influence insulin signaling via diverse mechanisms (Shoelson et al., 2006; Trujillo and Scherer, 2006) and these processes may interact with genetic variants influencing insulin resistance pathways (Manning et al., 2012). Adiposity levels have the potential to perturb the physiological milieu in which genetic variants in insulin signaling pathways operate (Manning et al., 2012). Therefore adiposity, and particularly fat in the pancreas and major glucose recipient organs, will exacerbate insulin resistance which consequently impairs beta cell function. Obesity also increases insulin demand therefore hyperfunction of beta cells may exhaust beta cells resulting in beta cell dysfunction.
By upregulating insulin secretion (Bergman, 2005) and the degree to which individuals are capable of achieving this determines whether or not an individual develops diabetes (Ashcroft and Rorsman, 2012). This, in turn, is influenced by their genetic constitution which explains why some obese and insulin resistant individuals do not develop type 2 diabetes; their beta cells can compensate (Ashcroft and Rorsman, 2012). In those individuals who develop diabetes, beta cell compensation declines due to hyperglycemia resulting in a persistent progressively increasing metabolic load that is influenced by factors such as aging and an increase in the number and severity of diabetogenic risk factors.
Saturated Fat and FFA Trigger Beta Cell Dysfunction and Insulin Resistance
Higher body mass indices (BMIs) may potentiate the effect of genetic variants on insulin resistance pathways, an effect that could be attributed to tissue-specific responses to the obesogenic environment (Prudente et al., 2009). The effects of FFA on beta cell function are time dependent. Short-term exposure to FFA increases GSIS which results in increased insulin secretion following a mixed meal and enables storage of excess calories as fat (Ashcroft and Rorsman, 2012) which contributes to overweight and obesity. FFA may account for the compensatory upregulation of beta cell function in response to insulin resistance (Stefanovski et al., 2011). Conversely, long-term exposure to FFA suppresses GSIS and has been suggested to involve impaired glucose metabolism, reduced insulin biosynthesis, and beta cell loss (Yaney and Corkey, 2003; Poitout and Robertson, 2008).
Obesity and high fat feeding mimic the effects of long-term incubation of islets with FFA on insulin secretion and Ca2+-channel distribution (Collins et al., 2010) which correlate with an increased amount of fat within the islets (Hoppa et al., 2009) and the surrounding exocrine pancreas (Pinnick et al., 2008). There is also an inverse correlation between the amount of fat in the human pancreas and GSIS, with glucose tolerance and insulin secretion improving in parallel with a reduction in pancreatic fat (Tushuizen et al., 2007). Intra-pancreatic or intra-islet fat depots may therefore provide a long-term local source of FFA that adversely affects beta cell function (Ashcroft and Rorsman, 2012). Obesity coupled to insulin resistance increases the functional demand per beta cell which would increase the burden and accelerate beta cell dysfunction. The pathogenesis of beta cell dysfunction may, to a certain extent, mimic hepatic steatosis: intra-tissue fat depots induce inflammation thereby triggering cellular demise and dysfunction. Intra-islet, in particular intra-beta cell, fat could therefore impair GSIS and insulin signaling in islets. This likely represents a mechanism for beta cell dysfunction and reduced beta cell compensation that impairs GSIS by non-glucose sensing by beta cells similar to impaired insulin signaling and consequently glucose uptake in glucose recipient tissues that triggers insulin resistance.
Islet inflammation (insulitis) in type 2 diabetes is attributed to nutrient overload leading to metabolic exhaustion in beta cells (Donath and Shoelson, 2011). This can lead to localized synthesis of cytokines, resulting in recruitment of immune cells to the site of production (Donath and Shoelson, 2011) which will trigger beta cell dysfunction and exacerbate insulin resistance. Nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) has a central regulatory role in FFA-induced inflammation and beta cell death (Choi et al., 2012). The saturated FFA, palmitate, induces beta cell dysfunction in vivo by activating inflammatory processes within mouse islets (Eguchi et al., 2012). Palmitate treatment increased expression of major cytokines implicated in beta cell dysfunction (Choi et al., 2012), viz., interleukin (IL) 6, IL8 (CXCL1), IP10 [chemokine (C-X-C motif) ligand 10 (CXCL10)], MCP1 (CCL2), and MIP1A (CCL3) (Donath et al., 2010), where they may influence beta cells in an autocrine manner (Choi et al., 2012). Insulin sensitivity is impaired by saturated FFA and improved by polyunsaturated FFA (Siri-Tarino et al., 2010). In rats, saturated FFA have shown to increase intramuscular palmitic acid accumulation that may lead to insulin resistance (Reynoso et al., 2003). In humans, a positive association between serum FFA composition and diabetes was reported (Vessby et al., 1994; Coelho et al., 2011).
Beta Cell Compensation
Upon beta cell demise, beta cell compensation occurs to restore beta cell physiology. Optimal control of blood glucose concentrations depends on subtle changes in insulin synthesis and secretion by beta cells and on their capacity for large increases in secretion after meals, requiring large stores of insulin (Tarabra et al., 2012). It is critical that islets maintain adequate beta cell mass in response to various fluctuations in demand (Tarabra et al., 2012). Beta cell mass is enhanced by proliferation (replication of beta cells), neogenesis (differentiation from non-beta cells), hyperplasia (increased beta cell number) and hypertrophy (increased beta cell size), and is decreased by beta cell death; through apoptosis, necrosis, autophagy, and ferroptosis; hypoplasia (decreased beta cell number); and hypotrophy (decreased beta cell size). The expansion and demise of beta cell mass through stimulants and insults respectively are likely triggered through one or more of these processes of beta cell replenishment (beta cell expansion) and death (beta cell demise).
Proliferation refers to an increase in beta cells from beta cell replication (beta cell self-replenishment) whereas beta cell hyperplasia occurs by beta cell replication or beta cell neogenesis from non-beta cells. Both beta cell replication and neogenesis contribute to the expansion of beta cell mass and require external stimuli such as hormones and growth factors (Bouwens and Rooman, 2005). Beta cells are dynamic and altered in response to fluctuating metabolic demand for insulin. Beta cell hypertrophy and hyperplasia occur during beta cell compensation to increase beta cell mass in response to hyperglycemia in diabetogenic states (Cerf et al., 2012). In several insulin resistant and diabetic rodent models, most islets were mildly enlarged and displayed hypertrophy and hyperplasia (Jones et al., 2010). Further, beta cell hypertrophy contributes to beta cell compensation in high fat diet-induced insulin resistance and the master beta cell transcription factor, pancreatic duodenal homeobox 1 (Pdx1), regulates beta cell size (Sachdeva et al., 2009), i.e., Pdx1 influences beta cell hyper- or hypotrophy. In contrast, beta cell hypotrophy results from beta cell death via various processes and insults and contributes to reduced beta cell mass. In addition, beta cell hypotrophy was found in hyperglycemic weanling rats exposed to a high fat diet during any single week of gestation (Cerf et al., 2007). Hyperglycemia may be exacerbated by the inability of hypotrophic and hypoplastic beta cells to synthesize and secrete sufficient insulin which consequently results in hypoinsulinemia (Cerf et al., 2007).
In diabetes, reduced beta cell mass occurs through apoptosis, necrosis, autophagy, and potentially ferroptosis. In human type 2 diabetes, both increased apoptosis and reduced replication may contribute to beta cell loss and reduced beta cell mass (Karaca et al., 2009). Beta cell hyperplasia and hyperinsulinemia compensate for progressively increasing insulin resistance to maintain normoglycemia; with time apoptosis exceeds the rate of replication and beta cell mass declines (Kiraly et al., 2008). The cytokine, IL1, induces beta cell necrosis suggesting that macrophage-derived cytokines participate in the initial pathogenesis of diabetes by inducing beta cell death by a mechanism that promotes necrosis and islet inflammation (Steer et al., 2006). Autophagy, a catabolic process that involves the degradation of cellular components through the lysosomal machinery, is important for maintaining normal islet homeostasis and compensatory beta cell hyperplasia in response to high fat dietary intake (Ebato et al., 2008). In type 2 diabetic patients, increased beta cell death was associated with altered autophagy suggesting that autophagy can be induced by metabolic perturbations (Marchetti and Masini, 2009). The cell death process of ferroptosis is morphologically, biochemically, and genetically distinct from apoptosis, various forms of necrosis and autophagy, and is characterized by iron-dependent accumulation of lethal lipid ROS (Dixon et al., 2012). The role of ferroptosis in beta cell demise still requires elucidation.
Beta cells initially compensate for the insulin resistance associated with obesity by increasing insulin secretion (Kasuga, 2006). When beta cell loss reaches the point of causing hyperglycemia, the beta cell replication rate is presumably maximally stimulated; therefore a further elevation in glucose concentrations will not increase replication (Porat et al., 2011). Glucose homeostasis maintains normoglycemia by adapting the mass and function of beta cells that counter insulin resistance, reduced beta cell mass, and excess nutrition (Liu et al., 2001).
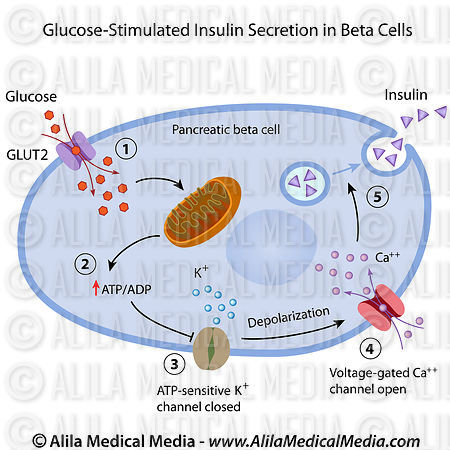
Glucose Potentiates Beta Cell Proliferation
The physiology of beta cells and insulin sensitive tissues are associated and mediated by metabolites, notably glucose. The beta cells compensate (adapt) in response to fluctuations in the demand for insulin. In states of increased insulin demand such as hyperglycemia, beta cells compensate to restore glucose homeostasis (maintain normoglycemia) via beta cell hypertrophy, and hyperplasia (to collectively increase beta cell mass) (Cerf, 2010) also increasing insulin biosynthesis (Brissova et al., 2005). Glucose increases the rate of beta cell proliferation in vitro (Kwon et al., 2004) and during short periods of glucose infusion in rodents (Bonner-Weir et al., 1989; Paris et al., 2003; Alonso et al., 2007) with glucose identified as the key systemic factor controlling beta cell replication (Porat et al., 2011). All beta cells appear to equally contribute to growth and maintenance (Brennand et al., 2007).
Glucose can increase the rate of beta cell proliferation in vitro (Kwon et al., 2004) and during short periods of glucose infusion in vivo (rodents) (Bonner-Weir et al., 1989; Paris et al., 2003; Alonso et al., 2007). Insulin, FFA, and incretin hormones have also been proposed as beta cell proliferative agents, particularly in insulin resistant states where circulating glucose concentrations are not measurably elevated but several other beta cell mitogens are yet to be identified (Porat et al., 2011).
A recent study identified a simple mechanism for homeostasis of beta cell proliferation and mass where beta cells adjust their proliferation rate according to the rate of glycolysis; this provides a system for sensitive measurement of organismal demand for beta cells, while normoglycemia is maintained (Porat et al., 2011). The same homeostatic mechanism appears to be responsible for the control of beta cell number during healthy adult life and during regeneration following injury (Porat et al., 2011). Physiologically, this model explains how beta cell number can be finely adjusted according to the organism’s needs without deviating from the normoglycemic range (Porat et al., 2011).
The capacity of beta cells to proliferate in response to insulin resistance is critical for glucose homeostasis and for preventing the progression of type 2 diabetes (Blandino-Rosano et al., 2012). Proliferation of mature beta cells is one of the components responsible for maintenance of beta cell mass in adult life (Dor et al., 2004). A highly perfused subpopulation of islets demonstrated a higher rate of beta cell proliferation (Lau et al., 2012). Islet endothelial paracrine factors can increase beta cell proliferation (Lau et al., 2012) and the transcription factor, Islet 1, has been identified as crucial for sustained beta cell proliferation and the prevention of apoptosis in postnatal beta cells (Du et al., 2009). Tcf7l2 promotes beta cell proliferation, protects against apoptosis, and improves insulin secretion (Loder et al., 2008; Shu et al., 2008, 2009).
Beta cell proliferation progressively reduces with age. Adaptive beta cell proliferation is severely restricted with advanced age (Rankin and Kushner, 2009). In mice, beta cell regeneration, which occurs mainly by self-renewal, is severely and abruptly restricted by middle age (Rankin and Kushner, 2009). The regenerative capacity of adult beta cells becomes limited by early middle age (1 year, about 40% of the life span in mice) (Harrison and Archer, 1987). Beta cell replication may become fully restricted when adult insulin requirements are established in middle age (Rankin and Kushner, 2009). In human islets from donors aged 17–74 years, there was an age-related decreased expression of Pdx1 indicating both a decreased capacity with age for cellular proliferation (decreased plasticity) and reduced insulin formation and secretion (Maedler et al., 2006). A significantly higher incidence of beta cell neogenesis was observed in the premature and developing human pancreas (Gregg et al., 2012). Postnatally, human beta cell neogenesis was observed at a negligible rate of ≤0.5% in normal human pancreata with several rare instances of beta cell neogenesis observed at 1, 15, and 39 years (Gregg et al., 2012).
Insulin secretion in the presence of resveratrol is increased in various pancreatic beta cell cultures (Chen et al., 2007).
Phenolic compounds potentially positively preserve beta cells and their function (Lin et al., 2012).
Quercetin and naringenin possibly protect beta cells from cytokine-induced toxicity by enhancing cell survival through the PI3K pathway, independent of p-p38 mitogen-activated protein kinases (MAPK), or inducible NO synthase (iNOS) as demonstrated in INS-1 cells (Lin et al., 2012).
In addition, quercetin and naringenin induce pAkt activation and could further participate in beta cell protection from cytokine-induced cell apoptosis, possibly by a NO-independent mechanism and could be potent agents to benefit beta cell mass preservation (Lin et al., 2012).
Healthy Diet and Regular Exercise
A healthy normocaloric diet that is well balanced, limited in saturated fat content, and meets the recommended daily allowances of key nutrients protects beta cells and enhances longevity in healthy individuals. Reduced energy intake combined with exercise improves insulin sensitivity (Weickert, 2012). Additional dietary measures that reduce insulin resistance include ingesting a Mediterranean dietary pattern that avoids excess dietary fat intake; substituting saturated fatty acids and trans fatty acids with monounsaturated fatty acids and polyunsaturated fatty acids, emphasizing cereal fiber content in the diet and when on a high protein diet, maintaining high levels of exercise (Weickert, 2012). Patients with poorly controlled type 2 diabetes who were placed on a hypocaloric diet for up to 12 weeks experienced a marked and rapid decrease in liver fat content (85%), associated specifically with normalization in hepatic insulin sensitivity and reductions in fasting hyperglycemia and hepatic glucose production without changes in intramyocellular lipid or insulin-mediated whole body glucose disposal (Petersen et al., 2005).
http://journal.frontiersin.org/article/10.3389/fendo.2013.00037/full
Regular exercise, although it may not necessarily reduce weight, enhances insulin sensitivity. Importantly, exercise concomitant with weight loss improves beta cell function (Dela et al., 2004; Solomon et al., 2010). Exercise maintained or enhanced beta cell function in older obese individuals, with improved beta cell function correlating with reduced glucose concentrations (Malin and Kirwan, 2012). In the ZDF rat, voluntary running prevented the development of diabetes despite continuing hyperphagia, obesity, and hyperlipidemia (Delghingaro-Augusto et al., 2012). Compensatory insulin secretion was preserved and hyperglycemia prevented by exercise was characterized by enhanced insulin secretion per islet and the prevention of severe depletion of islet insulin stores (Delghingaro-Augusto et al., 2012). Frequent exercise and adopting a healthy diet is a strategy to improve diabetic outcomes, particularly in pre-diabetic states such as obesity and/or insulin resistance and beta cell dysfunction.